Microbes that invade plant hosts, including fungal pathogens, use effectors to suppress, avoid, or mitigate the plant’s immune system. In turn, plants use a wide diversity of plasma membrane and cytoplasmically localized receptors to detect such invasion, either through recognition of the effector itself or its biochemical activity. Such detection leads to the activation of immune responses to arrest microbe ingress, and results in a strong selection pressure on the microbe to modify the effector or its function to alleviate recognition, in turn. In order to accommodate the genomic plasticity that is required to allow such modifications of effector catalogs in a swift manner, many plant-associated microbes evolved some form of compartmentalization within their genome, often referred to as a so-called “two-speed genome”. Typically, in such genome, a non-random genomic arrangement of effector genes occurs in repeat-rich regions that display signs of increased plasticity and accelerated evolution.
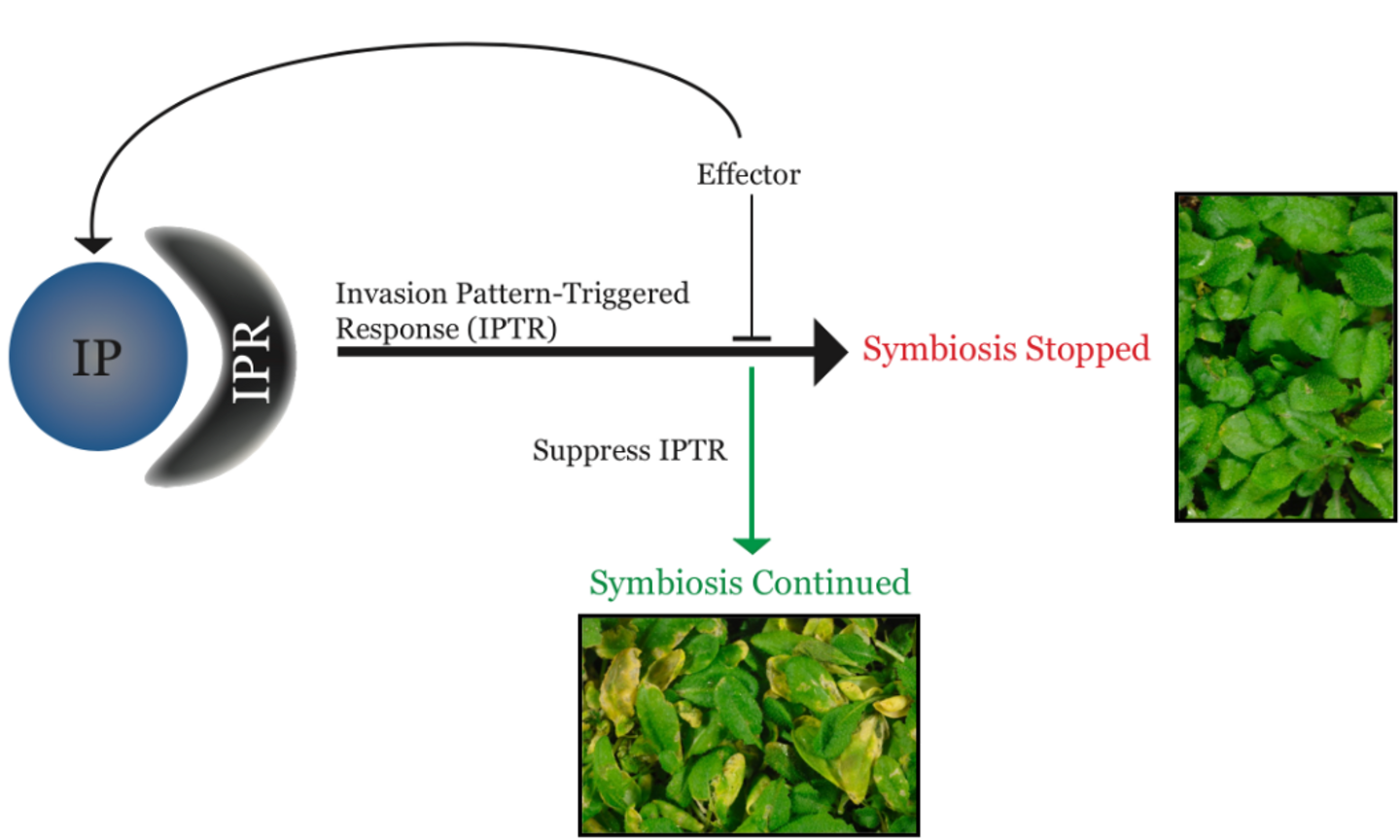
Plants use invasion pattern receptors to perceive invasion patterns, any type of molecule that can reliably betray invasion, to activate immune responses aimed at halting microbial ingress. Plant-associated microbes use effectors to perturb these immune responses in turn. However, effectors may become invasion patterns themselves again, once recognized by host immune receptors (figure adapted from Cook et al. 2016).
Verticillium dahliae presumably is a asexual Ascomycete fungus that generates genomic diversity in the absence of sexual recombination through large-scale chromosomal rearrangements along with the occurrence of segmental duplications. The regions that underwent such rearrangements are hypervariable between strains of the fungus, and have consequently been referred to as lineage-specific (LS) regions. These LS regions, initially defined solely based on presence/absence variation (PAV) between Verticillium dahliae strains are enriched for in planta expressed (effector) genes.
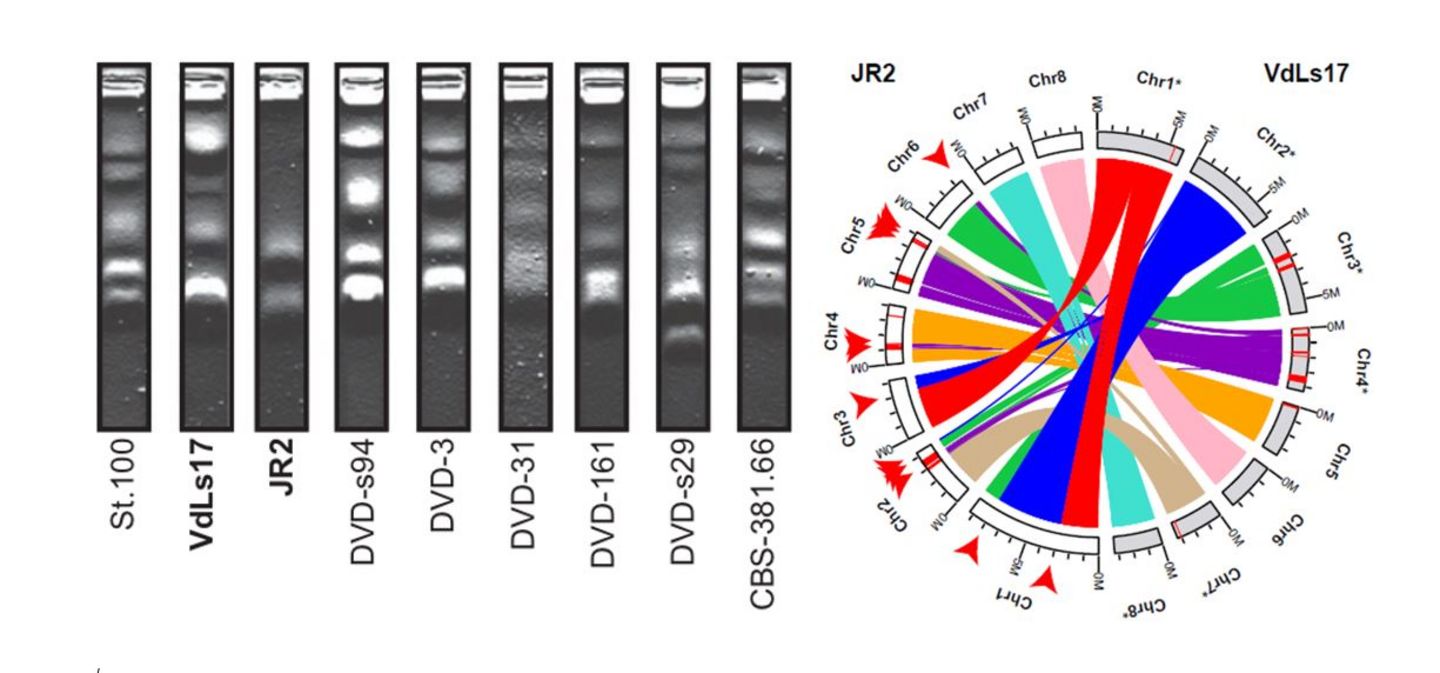
Left: Chromosomal karyotypes of a set of Verticillium dahliae strains as determined by pulsed-field gel electrophoresis, revealing significant chromosome length and structure polymorphisms between strains. Right: Circos diagram illustrating collinear blocks in alignments between the eight chromosomes of Verticillium dahliae strains VdLs17 (gray) and JR2 (white). Lineage-specific sequences are indicated as red segments on the chromosomes (figure adapted from de Jonge et al. 2013 & Faino et al., 2016).
Recently, more and more evidence in various filamentous plant-associated microbes point toward unexplained connections between genome organization, genomic variability and the epigenome, potentially with adaptive consequences. We recently demonstrated that the core genome of V. dahliae is organized into heterochromatic and euchromatic regions. In contrast to the euchromatin, the heterochromatin is characterized by a high transposable element (TE) density, low GC content, high levels of DNA and H3K9 methylation, low DNA accessibility and clear signatures of RIP mutations at repetitive sequences. Intriguingly, LS regions exist in a unique chromatin state: strongly associated with H3K27me3, despite their high TE density devoid of DNA and H3K9 methylation, with higher DNA accessibility than core heterochromatic regions and more transcriptionally active, but less accessible than euchromatic core regions. Notably, the chromatin state is akin to facultative heterochromatin.
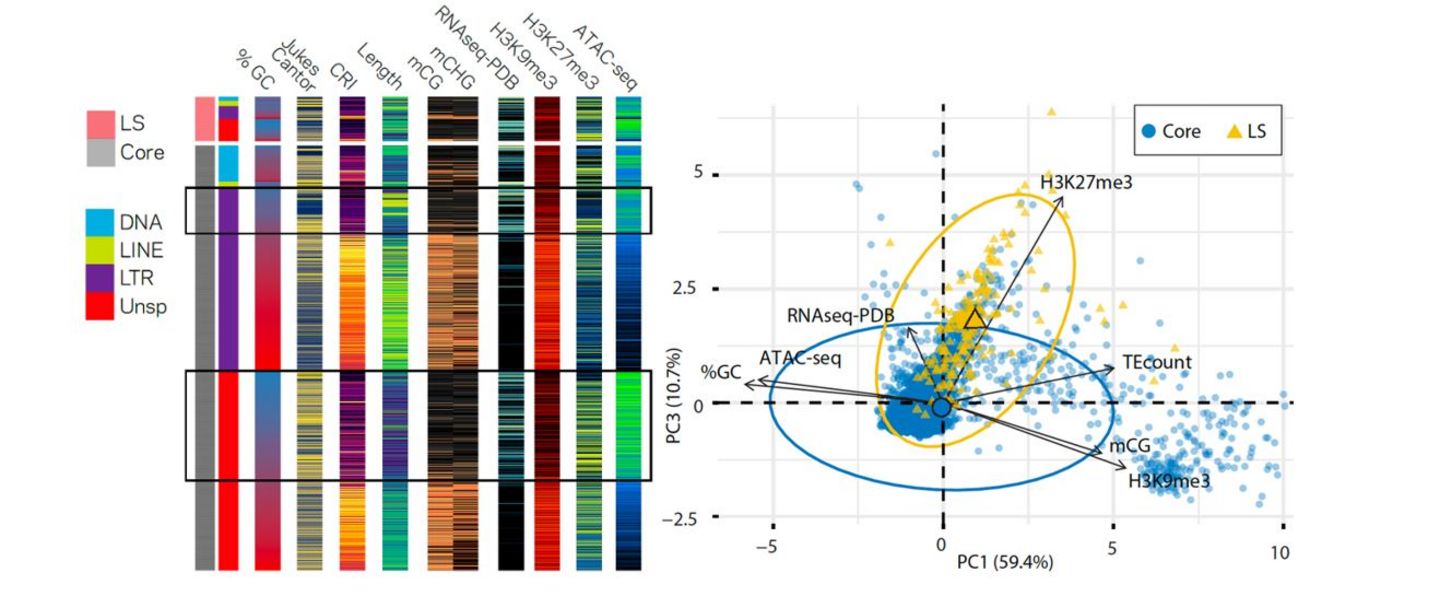
Left: Collective epigenome and physical DNA characteristics define core and LS regions in V. dahliae. Left: Grouped heatmaps for ten variables collected for transposable elements in the V. dahliae genome, arranged by LS (top) or core (below) localization. The boxes highlight core TEs with euchromatin profiles. Right: Principal component analysis for seven (epi)genomic variables in 10 kb windows either belonging to core or LS genomic regions (figure adapted from Cook et al. 2020).
Our epigenetic and physical DNA accessibility data suggest that genome structure influences genome function, and was used to predict LS regions in the genome of V. dahliae with machine learning. This analysis revealed twice as much LS DNA as originally recognized, which we propose to refer to as “adaptive genomic regions”. Arguably, the formation of adaptive genomic regions is not fully deterministic, as evolution is a stochastic and probabilistic process, in which specific chromatin and physical status increases the likelihood for adaptive traits on a continuum. Further experimental evidence is needed to determine how chromatin interacts with evolutionary forces to shape V. dahliae fitness.
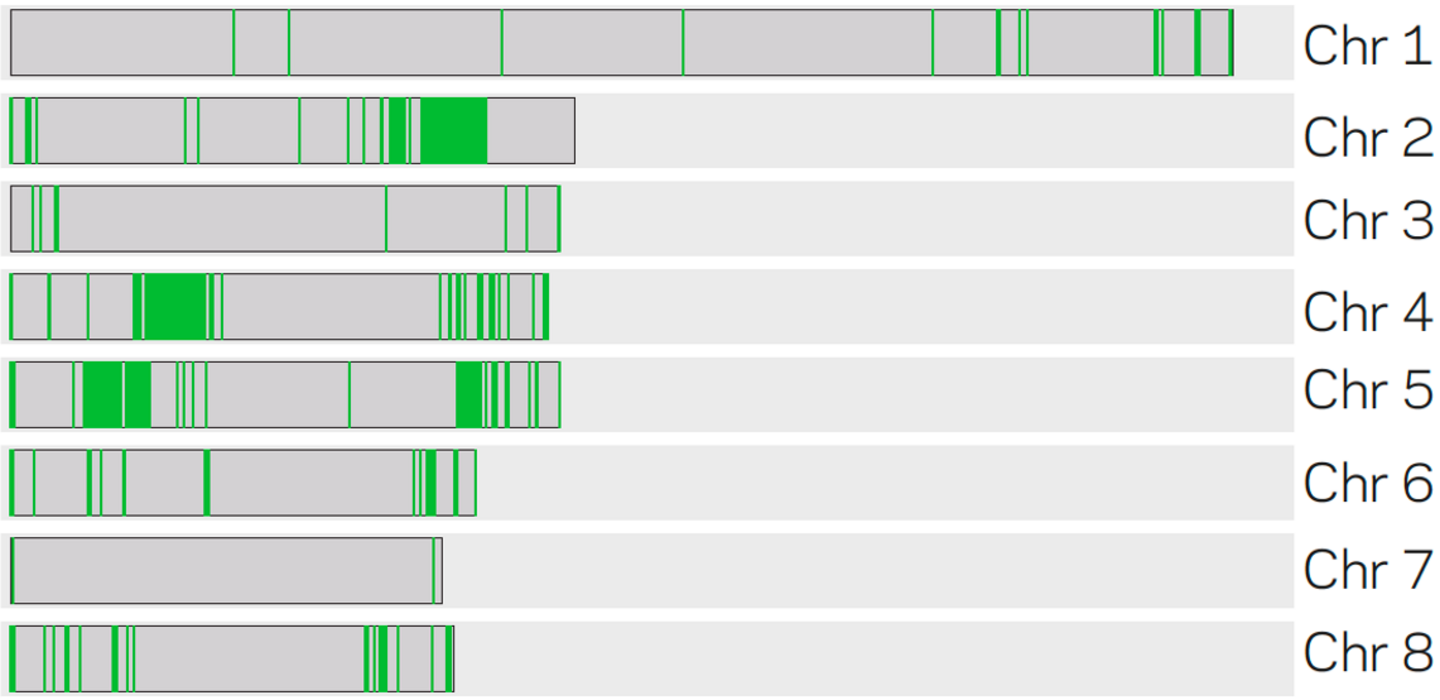
Schematic representation of core (gray) and adaptive (green) genomic regions on the eight chromosomesof V. dahliae strain JR2 based on machine learning (figure adapted from Cook et al. 2020).
Most relevant literature
Cook et al., (2020). A unique chromatin profile defines adaptive genomic regions in a fungal plant pathogen. eLife 9: e62208.
Seidl et al., (2020). Repetitive elements contribute to the diversity and evolution of centromeres in the fungal genus Verticillium. mBio 11: e01714-20.
Depotter et al. (2019). Dynamic virulence-related regions of the plant pathogenic fungus Verticillium dahliae display enhanced sequence conservation. Molecular Ecology 28: 3482-3495.
Shi-Kunne et al. (2018). Evolution within the fungal genus Verticillium is characterized by chromosomal rearrangement and gene loss. Environmental Microbiology 20: 1362-1373.
Seidl MF, Cook DE, Thomma BPHJ (2016). Chromatin biology impacts adaptive evolution of filamentous plant pathogens. PLoS Pathogens 12: e1005920.
Faino et al. (2016). Transposons passively and actively contribute to evolution of the two-speed genome of a fungal pathogen. Genome Research 26: 1091-1100.
Faino et al. (2015). Single-Molecule Real-Time sequencing combined with optical mapping yields completely finished fungal genome. mBio 6: e00936-15.
Cook DE, Mesarich CH, Thomma BPHJ (2015). Understanding plant immunity as a surveillance system to detect invasion. Annual Review of Phytopathology 53: 541-563.
Seidl & Thomma (2014). Sex or no sex: evolutionary adaptation occurs regardless. Bioessays 36: 335-345.
de Jonge et al. (2013). Extensive chromosomal reshuffling drives evolution of virulence in an asexual pathogen. Genome Research 23: 1271-1282.
de Jonge et al. (2012). Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proceedings of the National Academy of Sciences of the USA 109: 5110-5115.